Levitra Full Prescription Information
(vardenafil HCI) Tablets
Description
Pharmacology
Indications and Usage
Contraindications
Warnings
Precautions
Drug Interactions
Adverse Reactions
Overdose
Dosage
Supplied
DESCRIPTION
LEVITRA® is an oral therapy for the treatment of erectile dysfunction. This monohydrochloride salt of vardenafil is a selective inhibitor of cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5).
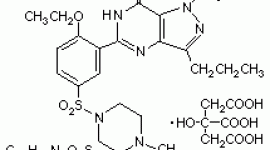
Vardenafil HCl is designated chemically as piperazine, 1-[[3-(1,4-dihydro-5- methyl-4-oxo-7-propylimidazo[5,1-f][1,2,4]triazin-2-yl)-4- ethoxyphenyl]sulfonyl]-4-ethyl-, monohydrochloride and has the following structural formula:

Vardenafil HCl is a nearly colorless, solid substance with a molecular weight of 579.1 g/mol and a solubility of 0.11 mg/mL in water. LEVITRA is formulated as orange, round, film-coated tablets with "BAYER" cross debossed on one side and "2.5", "5", "10", and "20" on the other side corresponding to 2.5 mg, 5 mg, 10 mg, and 20 mg of vardenafil, respectively. In addition to the active ingredient, vardenafil HCl, each tablet contains microcrystalline cellulose, crospovidone, colloidal silicon dioxide, magnesium stearate, hypromellose, polyethylene glycol, titanium dioxide, yellow ferric oxide, and red ferric oxide.
CLINICAL PHARMACOLOGY
Mechanism of Action
Penile erection is a hemodynamic process initiated by the relaxation of smooth muscle in the corpus cavernosum and its associated arterioles. During sexual stimulation, nitric oxide is released from nerve endings and endothelial cells in the corpus cavernosum. Nitric oxide activates the enzyme guanylate cyclase resulting in increased synthesis of cyclic guanosine monophosphate (cGMP) in the smooth muscle cells of the corpus cavernosum. The cGMP in turn triggers smooth muscle relaxation, allowing increased blood flow into the penis, resulting in erection. The tissue concentration of cGMP is regulated by both the rates of synthesis and degradation via phosphodiesterases (PDEs). The most abundant PDE in the human corpus cavernosum is the cGMPspecific phosphodiesterase type 5 (PDE5); therefore, the inhibition of PDE5 enhances erectile function by increasing the amount of cGMP. Because sexual stimulation is required to initiate the local release of nitric oxide, the inhibition of PDE5 has no effect in the absence of sexual stimulation. In vitro studies have shown that vardenafil is a selective inhibitor of PDE5. The inhibitory effect of vardenafil is more selective on PDE5 than for other known phosphodiesterases (>15-fold relative to PDE6, >130-fold relative to PDE1, >300-fold relative to PDE11, and >1,000-fold relative to PDE2, 3, 4, 7, 8, 9, and 10).
Pharmacokinetics
The pharmacokinetics of vardenafil are approximately dose proportional over the recommended dose range. Vardenafil is eliminated predominantly by hepatic metabolism, mainly by CYP3A4 and to a minor extent, CYP2C isoforms. Concomitant use with strong CYP3A4 inhibitors such as ritonavir, indinavir, ketoconazole, itraconazole as well as moderate CYP3A inhibitors such as erythromycin results in significant increases of plasma levels of vardenafil (see PRECAUTIONS, WARNINGS and DOSAGE AND ADMINISTRATION). Mean vardenafil plasma concentrations measured after the administration of a single oral dose of 20 mg to healthy male volunteers are depicted in Figure 1.
Figure 1: Plasma Vardenafil Concentration (Mean ± SD) Curve for a Single 20 mg LEVITRA Dose

Absorption: Vardenafil is rapidly absorbed with absolute bioavailability of approximately 15%. Maximum observed plasma concentrations after a single 20 mg dose in healthy volunteers are usually reached between 30 minutes and 2 hours (median 60 minutes) after oral dosing in the fasted state. Two foodeffect studies were conducted which showed that high-fat meals caused a reduction in Cmax by 18%-50%.
Distribution: The mean steady-state volume of distribution (Vss) for vardenafil is 208 L, indicating extensive tissue distribution. Vardenafil and its major circulating metabolite, M1, are highly bound to plasma proteins (about 95% for parent drug and M1). This protein binding is reversible and independent of total drug concentrations.
Following a single oral dose of 20 mg vardenafil in healthy volunteers, a mean of 0.00018% of the administered dose was obtained in semen 1.5 hours after dosing.
Metabolism: Vardenafil is metabolized predominantly by the hepatic enzyme CYP3A4, with contribution from the CYP3A5 and CYP2C isoforms. The major circulating metabolite, M1, results from desethylation at the piperazine moiety of vardenafil. M1 is subject to further metabolism. The plasma concentration of M1 is approximately 26% that of the parent compound. This metabolite shows a phosphodiesterase selectivity profile similar to that of vardenafil and an in vitro inhibitory potency for PDE5 28% of that of vardenafil. Therefore, M1 accounts for approximately 7% of total pharmacologic activity.
Excretion: The total body clearance of vardenafil is 56 L/h, and the terminal half-life of vardenafil and its primary metabolite (M1) is approximately 4-5 hours. After oral administration, vardenafil is excreted as metabolites predominantly in the feces (approximately 91-95% of administered oral dose) and to a lesser extent in the urine (approximately 2-6% of administered oral dose).
Pharmacokinetics in Special Populations
Pediatrics: Vardenafil trials were not conducted in the pediatric population.
Geriatrics: In a healthy volunteer study of elderly males (> 65 years) and younger males (18 - 45 years), mean Cmax and AUC were 34% and 52% higher, respectively, in the elderly males (see PRECAUTIONS, Geriatric Use and DOSAGE AND ADMINISTRATION). Consequently, a lower starting dose of LEVITRA (5 mg) in patients ≥ 65 years of age should be considered.
Renal Insufficiency: In volunteers with mild renal impairment (CLcr = 50-80 ml/min), the pharmacokinetics of vardenafil were similar to those observed in a control group with normal renal function. In the moderate (CLcr = 30-50 ml/min) or severe (CLcr 80 ml/min). Vardenafil pharmacokinetics have not been evaluated in patients requiring renal dialysis (see PRECAUTIONS, Renal Insufficiency, and DOSAGE AND ADMINISTRATION).
Hepatic Insufficiency: In volunteers with mild hepatic impairment (Child- Pugh A), the Cmax and AUC following a 10 mg vardenafil dose were increased by 22% and 17%, respectively, compared to healthy control subjects. In volunteers with moderate hepatic impairment (Child-Pugh B), the Cmax and AUC following a 10 mg vardenafil dose were increased by 130% and 160%, respectively, compared to healthy control subjects. Consequently, a starting dose of 5 mg is recommended for patients with moderate hepatic impairment, and the maximum dose should not exceed 10 mg (see PRECAUTIONS and DOSAGE AND ADMINISTRATION). Vardenafil has not been evaluated in patients with severe (Child-Pugh C) hepatic impairment.
Pharmacodynamics
Effects on Blood Pressure: In a clinical pharmacology study of patients with erectile dysfunction, single doses of vardenafil 20 mg caused a mean maximum decrease in supine blood pressure of 7 mm Hg systolic and 8 mm Hg diastolic (compared to placebo), accompanied by a mean maximum increase of heart rate of 4 beats per minute. The maximum decrease in blood pressure occurred between 1 and 4 hours after dosing. Following multiple dosing for 31 days, similar blood pressure responses were observed on Day 31 as on Day 1. Vardenafil may add to the blood pressure lowering effects of antihypertensive agents (see CONTRAINDICATIONS, PRECAUTIONS, Drug Interactions).
Effects on Blood Pressure and Heart Rate When LEVITRA is Combined with Nitrates: A study was conducted in which the blood pressure and heart rate response to 0.4 mg nitroglycerin (NTG) sublingually was evaluated in 18 healthy subjects following pretreatment with LEVITRA 20 mg at various times before NTG administration. LEVITRA 20 mg caused an additional time-related reduction in blood pressure and increase in heart rate in association with NTG administration. The blood pressure effects were observed when LEVITRA 20 mg was dosed 1 or 4 hours before NTG and the heart rate effects were observed when 20 mg was dosed 1, 4, or 8 hours before NTG. Additional blood pressure and heart rate changes were not detected when LEVITRA 20 mg was dosed 24 hours before NTG. (See Figure 2.)
Figure 2: Placebo-subtracted point estimates (with 90% CI) of mean maximal blood pressure and heart rate effects of pre-dosing with LEVITRA 20 mg at 24, 8, 4, and 1 hour before 0.4 mg NTG sublingually.

Because the disease state of patients requiring nitrate therapy is anticipated to increase the likelihood of hypotension, the use of vardenafil by patients on nitrate therapy or on nitric oxide donors is contraindicated (see CONTRAINDICATIONS).
Electrophysiology: The effect of 10 mg and 80 mg vardenafil on QT interval was evaluated in a single-dose, double-blind, randomized, placebo- and active-controlled (moxifloxacin 400 mg) crossover study in 59 healthy males (81% White, 12% Black, 7% Hispanic) aged 45-60 years. The QT interval was measured at one hour post dose because this time point approximates the average time of peak vardenafil concentration. The 80 mg dose of LEVITRA (four times the highest recommended dose) was chosen because this dose yields plasma concentrations covering those observed upon co-administration of a low-dose of LEVITRA (5 mg) and 600 mg BID of ritonavir. Of the CYP3A4 inhibitors that have been studied, ritonavir causes the most significant drug-drug interaction with vardenafil. Table 1 summarizes the effect on mean uncorrected QT and mean corrected QT interval (QTc) with different methods of correction (Fridericia and a linear individual correction method) at one hour post-dose. No single correction method is known to be more valid than the other. In this study, the mean increase in heart rate associated with a 10 mg dose of LEVITRA compared to placebo was 5 beats/minute and with an 80 mg dose of LEVITRA the mean increase was 6 beats/minute.
Table 1. Mean QT and QTc changes in msec (90% CI) from baseline relative to placebo at 1 hour post-dose with different methodologies to correct for the effect of heart rate.
| Table 1. Mean QT and QTc changes in msec (90% CI) from baseline relative to placebo at 1 hour post-dose with different methodologies to correct for the effect of heart rate. | |||
| Drug/Dose | QT Uncorrected (msec) | Fridericia QT Correction (msec) | Individual QT Correction (msec) |
| Vardenafil 10 mg | -2 (-4, 0) | 8 (6, 9) | 4 (3, 6) |
| Vardenafil 80 mg | -2 (-4, 0) | 10 (8, 11) | 6 (4, 7) |
| Moxifloxacin* 400 mg | 3 (1, 5) | 8 (6, 9) | 7 (5, 8) |
| * Active control (drug known to prolong QT) | |||
Therapeutic and supratherapeutic doses of vardenafil and the active control moxifloxacin produced similar increases in QTc interval. This study, however, was not designed to make direct statistical comparisons between the drugs or the dose levels. The actual clinical impact of these QTc changes is unknown. (See PRECAUTIONS).
Effects on Exercise Treadmill Test in Patients with Coronary Artery Disease (CAD): In two independent trials that assessed 10 mg (n=41) and 20 mg (n=39) vardenafil, respectively, vardenafil did not alter the total treadmill exercise time compared to placebo. The patient population included men aged 40-80 years with stable exercise-induced angina documented by at least one of the following: 1) prior history of MI, CABG, PTCA, or stenting (not within 6 months); 2) positive coronary angiogram showing at least 60% narrowing of the diameter of at least one major coronary artery; or 3) a positive stress echocardiogram or stress nuclear perfusion study.
Results of these studies showed that LEVITRA did not alter the total treadmill exercise time compared to placebo (10 mg LEVITRA vs. placebo: 433 ±109 and 426 ±105 seconds, respectively; 20 mg LEVITRA vs. placebo: 414 ±114 and 411 ±124 seconds, respectively). The total time to angina was not altered by LEVITRA when compared to placebo (10 mg LEVITRA vs. placebo: 291 ±123 and 292 ±110 seconds; 20 mg LEVITRA vs. placebo: 354 ±137 and 347 ±143 seconds, respectively). The total time to 1 mm or greater STsegment depression was similar to placebo in both the 10 mg and the 20 mg LEVITRA groups (10 mg LEVITRA vs. placebo: 380 ±108 and 334 ±108 seconds; 20 mg LEVITRA vs. placebo: 364 ±101 and 366 ±105 seconds, respectively).
Effects on Vision: Single oral doses of phosphodiesterase inhibitors have demonstrated transient dose-related impairment of color discrimination (blue/green) using the Farnsworth-Munsell 100-hue test and reductions in electroretinogram (ERG) b-wave amplitudes, with peak effects near the time of peak plasma levels. These finding are consistent with the inhibition of PDE6 in rods and cones, which is involved in phototransduction in the retina. The findings were most evident one hour after administration, diminishing but still present 6 hours after administration. In a single dose study in 25 normal males, LEVITRA 40 mg, twice the maximum daily recommended dose, did not alter visual acuity, intraocular pressure, fundoscopic and slit lamp findings.
CLINICAL STUDIES
Levitra was evaluated in four major double-blind, randomized, placebocontrolled, fixed-dose, parallel design, multi-center trials that enrolled 2431 men aged 20-83 (mean age 57 years; 78% White, 7% Black, 2% Asian, 3% Hispanic and 10% Other/Unknown). The doses of LEVITRA in these studies were 5 mg, 10 mg, and 20 mg. Two of these trials were conducted in the general ED population and two in special ED populations (one in patients with diabetes mellitus and one in post-prostatectomy patients). LEVITRA was dosed without regard to meals on an as needed basis in men with erectile dysfunction (ED), many of whom had multiple other medical conditions. The primary endpoints were assessed at 3 months.
Primary efficacy assessment in all four major trials was by means of the Erectile Function (EF) Domain score of the validated International Index of Erectile Function (IIEF) Questionnaire and two questions from the Sexual Encounter Profile (SEP) dealing with the ability to achieve vaginal penetration (SEP2), and the ability to maintain an erection long enough for successful intercourse (SEP3).
In all four fixed-dose efficacy trials, LEVITRA showed clinically meaningful and statistically significant improvement in the EF Domain, SEP2, and SEP3 scores compared to placebo. The mean baseline EF Domain score in these trials was 11.8 (scores range from 0-30 where lower scores represent more severe disease). LEVITRA (5 mg, 10 mg, and 20 mg) was effective in all age categories (<45, 45 to 65 years) and was also effective regardless of race (White, Black, Other).
Trials in a General Erectile Dysfunction Population: In the major North American fixed dose trial, 762 patients (mean age 57, range 20-83 years, 79% White, 13% Black, 4% Hispanic, 2% Asian and 2% Other) were evaluated. The mean baseline EF Domain scores were 13, 13, 13, 14 for the LEVITRA 5 mg, 10 mg, 20 mg and placebo groups, respectively. There was significant improvement (p<0.0001) at three months with LEVITRA (EF Domain scores of 18, 21, 21, for the 5 mg , 10 mg and 20 mg dose groups, respectively) compared to the placebo group (EF Domain score of 15). The European trial (total N=803) confirmed these results. The improvement in mean score was maintained at all doses at six months in the North American trial.
In the North American trial, LEVITRA significantly improved the rates of achieving an erection sufficient for penetration (SEP2) at doses of 5 mg, 10 mg, and 20 mg compared to placebo (65%, 75%, and 80%, respectively, compared to a 52% response in the placebo at 3 months; p< 0.0001). The European trial confirmed these results.
LEVITRA demonstrated a clinically meaningful and statistically significant increase in the overall per-patient rate of maintenance of erection to successful intercourse (SEP3) (51% on 5 mg, 64% on 10 mg, and 65% on 20 mg, respectively, compared to 32% on placebo, p< 0.0001) at 3 months in the North American trial. The European trial showed comparable efficacy. This improvement in mean score was maintained at all doses at 6 months in the North American trial.
Trial in Patients with ED and Diabetes Mellitus: LEVITRA demonstrated clinically meaningful and statistically significant improvement in erectile function in a prospective, fixed-dose (10 and 20 mg LEVITRA), double-blind, placebo-controlled trial of patients with diabetes mellitus (n=439; mean age 57 years, range 33-81; 80% White, 9% Black, 8% Hispanic, and 3% Other).
Significant improvements in the EF Domain were shown in this study (EF Domain scores of 17 on 10 mg LEVITRA and 19 on 20 mg LEVITRA compared to 13 on placebo; p< 0.0001).
LEVITRA significantly improved the overall per-patient rate of achieving an erection sufficient for penetration (SEP2) (61% on 10 mg and 64% on 20 mg LEVITRA compared to 36% on placebo; p< 0.0001).
LEVITRA demonstrated a clinically meaningful and statistically significant increase in the overall per-patient rate of maintenance of erection to successful intercourse (SEP3) (49% on 10 mg, 54% on 20 mg LEVITRA compared to 23% on placebo; p< 0.0001).
Trial in Patients with ED after Radical Prostatectomy: LEVITRA demonstrated clinically meaningful and statistically significant improvement in erectile function in a prospective, fixed-dose (10 and 20 mg LEVITRA), double-blind, placebo-controlled trial in post-prostatectomy patients (n=427, mean age 60, range 44-77 years; 93% White, 5% Black, 2% Other).
Significant improvements in the EF Domain were shown in this study (EF Domain scores of 15 on 10 mg LEVITRA and 15 on 20 mg LEVITRA compared to 9 on placebo; p< 0.0001).
LEVITRA significantly improved the overall per-patient rate of achieving an erection sufficient for penetration (SEP2) (47% on 10 mg and 48% on 20 mg LEVITRA compared to 22% on placebo; p <0.0001).
LEVITRA demonstrated a clinically meaningful and statistically significant increase in the overall per-patient rate of maintenance of erection to successful intercourse (SEP3) (37% on 10 mg, 34% on 20 mg LEVITRA compared to 10% on placebo; p< 0.0001).
INDICATIONS AND USAGE
LEVITRA is indicated for the treatment of erectile dysfunction.
CONTRAINDICATIONS
Nitrates: Administration of LEVITRA with nitrates (either regularly and/or intermittently) and nitric oxide donors is contraindicated (see CLINICAL PHARMACOLOGY, Pharmacodynamics, Effects on Blood Pressure and Heart Rate when LEVITRA is Combined with Nitrates). Consistent with the effects of PDE5 inhibition on the nitric oxide/cyclic guanosine monophosphate pathway, PDE5 inhibitors may potentiate the hypotensive effects of nitrates. A suitable time interval following LEVITRA dosing for the safe administration of nitrates or nitric oxide donors has not been determined.
Alpha Blockers: Because the co-administration of alpha-blockers and LEVITRA can produce hypotension, LEVITRA is contraindicated in patients taking alpha-blockers (see PRECAUTIONS, Drug Interactions).
Hypersensitivity: LEVITRA is contraindicated for patients with a known hypersensitivity to any component of the tablet.
WARNINGS
Cardiovascular effects
General: Physicians should consider the cardiovascular status of their patients, since there is a degree of cardiac risk associated with sexual activity. In men for whom sexual activity is not recommended because of their underlying cardiovascular status, any treatment for erectile dysfunction, including LEVITRA, generally should not be used.
Left Ventricular Outflow Obstruction: Patients with left ventricular outflow obstruction, e.g., aortic stenosis and idiopathic hypertrophic subaortic stenosis, can be sensitive to the action of vasodilators including Type 5 phosphodiesterase inhibitors.
Blood Pressure Effects: LEVITRA has systemic vasodilatory properties that resulted in transient decreases in supine blood pressure in healthy volunteers (mean maximum decrease of 7 mmHg systolic and 8 mmHg diastolic) (see CLINICAL PHARMACOLOGY, Pharmacodynamics). While this normally would be expected to be of little consequence in most patients, prior to prescribing LEVITRA, physicians should carefully consider whether their patients with underlying cardiovascular disease could be affected adversely by such vasodilatory effects.
Effect of Co-administration of Strong CYP3A4 inhibitors
Long-term safety information is not available on the concomitant administration of vardenafil with HIV protease inhibitors. Concomitant administration with ritonavir or indinavir substantially increases plasma concentrations of vardenafil. To decrease the chance of adverse events in patients concomitantly taking ritonavir or indinavir, which are strong inhibitors of CYP3A4 metabolism, a maximum single dose of 2.5 mg LEVITRA should not be exceeded. Because ritonavir prolongs LEVITRA elimination half-life (5-6-fold), no more than a single 2.5 mg dose of LEVITRA should be taken in a 72-hour period by patients also taking ritonavir. Patients taking indinavir, ketoconazole 400 mg daily, or itraconazole 400 mg daily should not exceed LEVITRA 2.5 mg once daily. For patients taking ketoconazole or itraconazole 200 mg daily, a single dose of 5 mg LEVITRA should not be exceeded in a 24-hour period (see PRECAUTIONS, Drug Interactions and DOSAGE AND ADMINISTRATION).
Other Effects
There have been rare reports of prolonged erections greater than 4 hours and priapism (painful erections greater than 6 hours in duration) for this class of compounds, including vardenafil. In the event that an erection persists longer than 4 hours, the patient should seek immediate medical assistance. If priapism is not treated immediately, penile tissue damage and permanent loss of potency may result.
Patient Subgroups Not Studied in Clinical Trials
There is no controlled clinical data on the safety or efficacy of LEVITRA in the following patients; and therefore its use is not recommended until further information is available.
- unstable angina; hypotension (resting systolic blood pressure of 170/110 mm Hg); recent history of stroke, life-threatening arrhythmia, or myocardial infarction (within the last 6 months); severe cardiac failure - severe hepatic impairment (Child-Pugh C) - end stage renal disease requiring dialysis - known hereditary degenerative retinal disorders, including retinitis pigmentosa
PRECAUTIONS
The evaluation of erectile dysfunction should include a determination of potential underlying causes, a medical assessment, and the identification of appropriate treatment.
Before prescribing LEVITRA, it is important to note the following:
Alpha-blockers: Caution is advised when PDE5 inhibitors are co-administered with alpha-blockers. Phosphodiesterase Type 5 (PDE5) inhibitors, including LEVITRA, and alpha-adrenergic blocking agents are both vasosdilators with blood-pressure lowering effects. When vasodilators are used in combination, an additive effect on blood pressure may be anticipated. In some patients, concomitant use of these two drug classes can lower blood pressure significantly (see PRECAUTIONS, Drug Interactions) leading to symptomatic hypotension (e.g., fainting). Consideration should be given to the following:
- Patients should be stable on alpha-blocker therapy prior to initiating a PDE5 inhibitor. Patients who demonstrate hemodynamic instability on alpha-blocker therapy alone are at increased risk of symptomatic hypotension with concomitant use of PDE5 inhibitors.
- In those patients who are stable on alpha-blocker therapy, PDE5 inhibitors should be initiated at the lowest recommended starting dose (see DOSAGE and ADMINISTRATION).
- In those patients already taking an optimized dose of PDE5 inhibitor, alpha-blocker therapy should be initiated at the lowest dose. Stepwise increase in alpha-blocker dose may be associated with further lowering of blood pressure in patients taking a PDE5 inhibitor.
- Safety of combined use of PDE5 inhibitors and alpha-blockers may be affected by other variables, including intravascular volume depletion and other anti-hypertensive drugs.
Hepatic Insufficiency: In volunteers with moderate impairment (Child-Pugh B), the Cmax and AUC following a 10 mg vardenafil dose were increased 130% and 160%, respectively, compared to healthy control subjects. Consequently, a starting dose of 5 mg is recommended for patients with moderate hepatic impairment and the maximum dose should not exceed 10 mg (see CLINICAL PHARMACOLOGY, Pharmacokinetics in Special Populations, and DOSAGE AND ADMINISTRATION). Vardenafil has not been evaluated in patients with severe hepatic impairment (Child-Pugh C).
Congenital or Acquired QT Prolongation: In a study of the effect of LEVITRA on QT interval in 59 healthy males (see CLINICAL PHARMACOLOGY, Electrophysiology), therapeutic (10 mg) and supratherapeutic (80 mg) doses of LEVITRA and the active control moxifloxacin (400 mg) produced similar increases in QTc interval. This observation should be considered in clinical decisions when prescribing LEVITRA. Patients with congenital QT prolongation and those taking Class IA (e.g., quinidine, procainamide) or Class III (e.g.,amiodarone, sotalol) antiarrhythmic medications should avoid using LEVITRA.
Renal Insufficiency: In patients with moderate (CLcr = 30-50 ml/min) to severe (CLcr 80 ml/min) (see CLINICAL PHARMACOLOGY, Pharmacokinetics in Special Populations). Vardenafil pharmacokinetics have not been evaluated in patients requiring renal dialysis.
General: In humans, vardenafil alone in doses up to 20 mg does not prolong the bleeding time. There is no clinical evidence of any additive prolongation of the bleeding time when vardenafil is administered with aspirin. Vardenafil has not been administered to patients with bleeding disorders or significant active peptic ulceration. Therefore LEVITRA should be administered to these patients after careful benefit-risk assessment.
Treatment for erectile dysfunction should generally be used with caution by patients with anatomical deformation of the penis (such as angulation, cavernosal fibrosis, or Peyronie's disease) or by patients who have conditions that may predispose them to priapism (such as sickle cell anemia, multiple myeloma, or leukemia).
The safety and efficacy of LEVITRA used in combination with other treatments for erectile dysfunction have not been studied. Therefore, the use of such combinations is not recommended.
Information for Patients
Physicians should discuss with patients the contraindication of LEVITRA with regular and/or intermittent use of organic nitrates. Patients should be counseled that concomitant use of LEVITRA with nitrates could cause blood pressure to suddenly drop to an unsafe level, resulting in dizziness, syncope, or even heart attack or stroke.
Physicians should inform their patients that concomitant use of LEVITRA with alpha-blockers is contraindicated because co-administration can produce hypotension (e.g. fainting). Patients prescribed LEVITRA who are taking alpha-blockers should be started on the lowest recommended starting dose of LEVITRA (see Drug Interactiona and DOSAGE AND ADMINISTRATION). Patients should be advised of the possible occurrence of symptoms related to postural hypotension and appropriate countermeasures. Patients should be advised to contact the prescribing physician if other anti-hypertensive drugs or new medications that may interact with LEVITRA are prescribed by another healthcare provider.
Physicians should advise patients to stop use of all PDE5 inhibitors, including LEVITRA, and seek medical attention in the event of sudden loss of vision in one or both eyes. Such an event may be a sign of non-arteritic anterior ischemic optic neuropathy (NAION), a cause of decreased vision, including permanent loss of vision, that has been reported rarely post-marketing in temporal association with the use of all PDE5 inhibitors. It is not possible to determine whether these events were related directly to the use of PDE5 inhibitors or to other factors. Physicians should also discuss with patients the increased risk of NAION in individuals who have already experienced NAION in one eye, including whether such individuals could be adversely affected by use of vasodilators such as PDE5 inhibitors (see POST-MARKETING EXPERIENCE/Ophthalmologic).
Physicians should discuss with patients the potential cardiac risk of sexual activity for patients with preexisting cardiovascular risk factors.
The use of LEVITRA offers no protection against sexually transmitted diseases. Counseling of patients about protective measures necessary to guard against sexually transmitted diseases, including the Human Immunodeficiency Virus (HIV), should be considered.
Physicians should inform patients that there have been rare reports of prolonged erections greater than 4 hours and priapism (painful erections greater than 6 hours in duration) for LEVITRA and this class of compounds. In the event that an erection persists longer than 4 hours, the patient should seek immediate medical assistance. If priapism is not treated immediately, penile tissue damage and permanent loss of potency may result.
Drug Interactions
Effect of other drugs on LEVITRA
In vitro studies: Studies in human liver microsomes showed that vardenafil is metabolized primarily by cytochrome P450 (CYP) isoforms 3A4/5, and to a lesser degree by CYP 2C9. Therefore, inhibitors of these enzymes are expected to reduce vardenafil clearance (see WARNINGS and DOSAGE AND ADMINISTRATION).
In vivo studies: Cytochrome P450 Inhibitors
Cimetidine (400 mg b.i.d.) had no effect on vardenafil bioavailability (AUC) and maximum concentration (Cmax) of vardenafil when co-administered with 20 mg LEVITRA in healthy volunteers. Erythromycin (500 mg t.i.d) produced a 4-fold increase in vardenafil AUC and a 3-fold increase in Cmax when co-administered with LEVITRA 5 mg in healthy volunteers (see DOSAGE AND ADMINISTRATION). It is recommended not to exceed a single 5 mg dose of LEVITRA in a 24-hour period when used in combination with erythromycin.
Ketoconazole (200 mg once daily) produced a 10-fold increase in vardenafil AUC and a 4-fold increase in Cmax when co-administered with LEVITRA (5 mg) in healthy volunteers. A 5-mg LEVITRA dose should not be exceeded when used in combination with 200 mg once daily ketoconazole. Since higher doses of ketoconazole (400 mg daily) may result in higher increases in Cmax and AUC, a single 2.5 mg dose of LEVITRA should not be exceeded in a 24-hour period when used in combination with ketoconazole 400 mg daily (see WARNINGS and DOSAGE AND ADMINISTRATION).
HIV Protease Inhibitors:
Indinavir (800 mg t.i.d.) co-administered with LEVITRA 10 mg resulted in a 16-fold increase in vardenafil AUC, a 7-fold increase in vardenafil Cmax and a 2-fold increase in vardenafil half-life. It is recommended not to exceed a single 2.5 mg LEVITRA dose in a 24-hour period when used in combination with indinavir (see WARNINGS and DOSAGE AND ADMINISTRATION).
Ritonavir (600 mg b.i.d.) co-administered with LEVITRA 5 mg resulted in a 49-fold increase in vardenafil AUC and a 13-fold increase in vardenafil Cmax. The interaction is a consequence of blocking hepatic metabolism of vardenafil by ritonavir, a highly potent CYP3A4 inhibitor, which also inhibits CYP2C9. Ritonavir significantly prolonged the half-life of vardenafil to 26 hours. Consequently, it is recommended not to exceed a single 2.5 mg LEVITRA dose in a 72-hour period when used in combination with ritonavir (see WARNINGS and DOSAGE AND ADMINISTRATION).
Other Drug Interactions: No pharmacokinetic interactions were observed between vardenafil and the following drugs: glyburide, warfarin, digoxin, Maalox, and ranitidine. In the warfarin study, vardenafil had no effect on the prothrombin time or other pharmacodynamic parameters.
Effects of LEVITRA on other drugs
In vitro studies:
Vardenafil and its metabolites had no effect on CYP1A2, 2A6, and 2E1 (Ki > 100μM). Weak inhibitory effects toward other isoforms (CYP2C8, 2C9, 2C19, 2D6, 3A4) were found, but Ki values were in excess of plasma concentrations achieved following dosing. The most potent inhibitory activity was observed for vardenafil metabolite M1, which had a Ki of 1.4 μM) toward CYP3A4, which is about 20 times higher than the M1 Cmax values after an 80 mg LEVITRA dose.
In vivo studies:
Nitrates: The blood pressure lowering effects of sublingual nitrates (0.4 mg) taken 1 and 4 hours after vardenafil and increases in heart rate when taken at 1, 4 and 8 hours were potentiated by a 20 mg dose of LEVITRA in healthy middle-aged subjects. These effects were not observed when LEVITRA 20 mg was taken 24 hours before the NTG. Potentiation of the hypotensive effects of nitrates for patients with ischemic heart disease has not been evaluated, and concomitant use of LEVITRA and nitrates is contraindicated (see CLINICAL PHARMACOLOGY, Pharmacodynamics, Effects on Blood Pressure and Heart Rate When LEVITRA is Combined with Nitrates; CONTRAINDICATIONS).
Nifedipine: Vardenafil 20 mg, when co-administered with slow-release nifedipine 30 mg or 60 mg once daily, did not affect the relative bioavailability (AUC) or maximum concentration (Cmax) of nifedipine, a drug that is metabolized via CYP3A4. Nifedipine did not alter the plasma levels of LEVITRA when taken in combination. In these patients whose hypertension was controlled with nifedipine, LEVITRA 20 mg produced mean additional supine systolic/diastolic blood pressure reductions of 6/5 mm Hg compared to placebo.
Alpha-blockers:
Blood pressure effects in patients on stable alpha-blocker treatment: Two clinical pharmacology studies were conducted in patients with benign prostatic hyperplasia (BPH) on stable-dose alpha-blocker treatment for at least four weeks.
Study 1: This study was designed to evaluate the effect of 5 mg vardenafil compared to placebo when administered to BPH patients on chronic alpha-blocker therapy in two separate cohorts: tamsulosin 0.4 mg daily (cohort 1, n=21) and terazosin 5 or 10 mg daily (cohort 2, n=21). The design was a randomized, double blind, cross-over study with four treatments: vardenafil 5 mg or placebo administered simultaneously with the alpha-blocker and vardenafil 5 mg or placebo administered 6 hours after the alpha-blocker. Blood pressure and pulse were evaluated over the 6-hour interval after vardenafil dosing. For BP results see Table 2. One patient after simultaneous treatment with 5 mg vardenafil and 10 mg terazosin exhibited symptomatic hypotension with standing blood pressure of 80/60 mmHg occurring one hour after administration and subsequent mild dizziness and moderate lightheadedness lasting for 6 hours. For vardenafil and placebo, five and two patients, respectively, experienced a decrease in standing systolic blood pressure (SBP) of >30 mmHg following simultaneous administration of terazosin. Hypotension was not observed when vardenafil 5 mg and terazosin were administered 6 hours apart. Following simultaneous administration of vardenafil 5 mg and tamsulosin, two patients had a standing SBP of 30 mmHg. When tamsulosin and vardenafil 5 mg were separated by 6 hours, two patients had a standing SBP 30 mmHg. There were no severe adverse events related to hypotension reported during the study. There were no cases of syncope.
Table 2: Mean (95% C.I.) maximal change from baseline in systolic blood pressure (mmH following vardenafil 5 mg in BPH patients on stable alpha-blocker therapy (Study 1)
| Alpha-Blocker | Simultaneous dosing of Vardenafil 5 mg and Alpha-Blocker, Placebo-Subtracted | Dosing of Vardenafil 5 mg and Alpha-Blocker Separated by 6 Hours, Placebo-Subtracted | |
| Terazosin 5 or 10 mg daily | Standing SBP | -3 (-6.7, 0.1) | -4 (-7.4, -0.5) |
| Supine SBP | -4 (-6.7, -0.5) | -4 (-7.1, -0.7) | |
| Tamsulosin 0.4 mg daily | Standing SBP | -6 (-9.9, -2.1) | -4 (-8.3, -0.5) |
| Supine SBP | -4 (-7.0, -0.8) | -5 (-7.9, -1.7) |
Study 2: This study was designed to evaluate the effect of 10 mg vardenafil (stage 1) and 20 mg vardenafil (stage 2) compared to placebo, when administered to a single cohort of BPH patients (n=23) on stable therapy with tamsulosin 0.4 mg or 0.8 mg daily for at least four weeks. The design was a randomized, double blind, two-period cross-over study. Vardenafil or placebo was given simultaneously with tamsulosin. Blood pressure and pulse were evaluated over the 6-hour interval after vardenafil dosing. For BP results see Table 3. One patient experienced a decrease from baseline in standing SBP of >30 mmHg following vardenafil 10 mg. There were no other instances of outlier blood pressure values (standing SBP 30 mmHg). Three patients reported dizziness following vardenafil 20 mg. There were no cases of syncope.
Table 3: Mean (95% C.I.) maximal change from baseline in systolic blood pressure (mmHg) following vardenafil 10 and 20 mg in BPH patients on stable alpha-blocker therapy with tamsulosin 0.4 or 0.8 mg daily (Study 2)
| Vardenafil 10 mg Placebo-subtracted | Vardenafil 20 mg Placebo-subtracted | |
| Standing SBP | -4 (-6.8, -0.3) | -4 (-6.8, -1.4) |
| Supine SBP | -5 (-8.2, -0.8) | -4 (-6.3, -1.8) |
Concomitant treatment with vardenafil and alpha-blockers should be initiated only if the patient is stable on his alpha-blocker therapy. In those patients who are stable on alpha-blocker therapy, LEVITRA should be initiated at the lowest recommended starting dose (see DOSAGE and ADMINISTRATION).
Blood pressure effects in normotensive men after forced titration with alpha-blockers:
Two randomized, double blind, placebo-controlled clinical pharmacology studies with healthy normotensive volunteers (age range, 45-74 years) were performed after forced titration of the alphablocker terazosin to 10 mg daily over 14 days (n=29), and after initiation of tamsulosin 0.4 mg daily for five days (n=24). There were no severe adverse events related to hypotension in either study. Symptoms of hypotension were a cause for withdrawal in 2 subjects receiving terazosin and in 4 subjects receiving tamsulosin. Instances of outlier blood pressure values (defined as standing SBP 30 mmHg) were observed in 9/24 subjects receiving tamsulosin and 19/29 receiving terazosin. The incidence of subjects with standing SBP <85 mmHg given vardenafil and terazosin to achieve simultaneous Tmax led to early termination of that arm of the study. In most (7/8) of these subjects, instances of standing SBP <85 mmHg were not associated with symptoms. Among subjects treated with terazosin, outlier values were observed more frequently when vardenafil and terazosin were given to achieve simultaneous Tmax than when dosing was administered to separate Tmax by 6 hours. There were 3 cases of dizziness observed with concomitant administration of terazosin and vardenafil. Seven subjects experienced dizziness mainly occurring with simultaneous Tmax administration of tamsulosin. There were no cases of syncope.
Table 4. Mean (95% C.I.) maximal change in baseline in systolic blood pressure (mmHg) following vardenafil 10 and 20 mg in healthy volunteers on daily alpha-blocker therapy
| Dosing of Vardenafil and Alpha-Blocker Separated by 6 Hours | Simultaneous dosing of Vardenafil and Alpha-Blocker | ||||
| Alphablocker | Vardenafil 10 mg Placebo- Subtracted | Vardenafil 20 mg Placebo- Subtracted | Vardenafil 10 mg Placebo- Subtracted | Vardenafil 20 mg Placebo- Subtracted | |
| Terazosin 10 mg daily | Standing SBP | -7 (-10, -3) | -11 (-14, -7) | -23 (-31, 16)* | -14 (-33, 11)* |
| Supine SBP | -5 (-8, -2) | -7 (-11, -4) | -7 (-25, 19)* | -7 (-31, 22)* | |
| Tamsulosi n 0.4 mg daily | Standing SBP | -4 (-8, -1) | -8 (-11, -4) | -8 (-14, -2) | -8 (-14, -1) |
| Supine SBP | -4 (-8, 0) | -7 (-11, -3) | -5 (-9, -2) | -3 (-7, 0) | |
* Due to the sample size, confidence intervals may not be an accurate measure for these data. These values represent the range for the difference.
Figure 6: Mean change from baseline in standing systolic blood pressure (mmHg) over 6 hour interval following simultaneous or 6 hr separation administration of vardenafil 10 mg, vardenafil 20 mg or placebo with terazosin (10 mg) in healthy volunteers

Figure 7: Mean change from baseline in standing systolic blood pressure (mmHg) over 6 hour interval following simultaneous or 6 hr separation administration of vardenafil 10 mg, vardenafil 20 mg or placebo with tamsulosin (0.4 mg) in healthy volunteers

Ritonavir and Indinavir: Upon concomitant administration of 5 mg of LEVITRA with 600 mg BID ritonavir, the Cmax and AUC of ritonavir were reduced by approximately 20%. Upon administration of 10 mg of LEVITRA with 800 mg TID indinavir , the Cmax and AUC of indinavir were reduced by 40% and 30%, respectively.
Alcohol: Alcohol (0.5 g/kg body weight: approximately 40 mL of absolute alcohol in a 70 kg person) and vardenafil plasma levels were not altered when dosed simultaneously. LEVITRA (20 mg) did not potentiate the hypotensive effects of alcohol during the 4-hour observation period in healthy volunteers when administered with alcohol (0.5 g/kg body weight).
Aspirin: LEVITRA (10 mg and 20 mg) did not potentiate the increase in bleeding time caused by aspirin (two 81 mg tablets).
Other interactions: LEVITRA had no effect on the pharmacodynamics of glyburide (glucose and insulin concentrations) and warfarin (prothrombin time or other pharmacodynamic parameters).
Carcinogenesis, Mutagenesis, Impairment of Fertility
Vardenafil was not carcinogenic in rats and mice when administered daily for 24 months. In these studies systemic drug exposures (AUCs) for unbound (free) vardenafil and its major metabolite were approximately 400- and 170- fold for male and female rats, respectively, and 21- and 37-fold for male and female mice, respectively, the exposures observed in human males given the Maximum Recommended Human Dose (MRHD) of 20 mg. Vardenafil was not mutagenic as assessed in either the in vitro bacterial Ames assay or the forward mutation assay in Chinese hamster V79 cells. Vardenafil was not clastogenic as assessed in either the in vitro chromosomal aberration test or the in vivo mouse micronucleus test. Vardenafil did not impair fertility in male and female rats administered doses up to 100 mg/kg/day for 28 days prior to mating in male, and for 14 days prior to mating and through day 7 of gestation in females. In a corresponding 1-month rat toxicity study, this dose produced an AUC value for unbound vardenafil 200 fold greater than AUC in humans at the MRHD of 20 mg.
There was no effect on sperm motility or morphology after single 20 mg oral doses of vardenafil in healthy volunteers.
Pregnancy, Nursing Mothers and Pediatric Use
LEVITRA is not indicated for use in women, newborns, or children. Vardenafil was secreted into the milk of lactating rats at concentrations approximately 10-fold greater than found in the plasma. Following a single oral dose of 3 mg/kg, 3.3% of the administered dose was excreted into the milk within 24 hours. It is not known if vardenafil is excreted in human breast milk.
Pregnancy Category B: No evidence of specific potential for teratogenicity, embryotoxicity or fetotoxicity was observed in rats and rabbits that received vardenafil at up to 18 mg/kg/day during organogenesis. This dose is approximately 100 fold (rat) and 29 fold (rabbit) greater than the AUC values for unbound vardenafil and its major metabolite in humans given the MRHD of 20 mg. In the rat pre-and postnatal development study, the NOAEL (no observed adverse effect level) for maternal toxicity was 8 mg/kg/day. Retarded physical development of pups in the absence of maternal effects was observed following maternal exposure to 1 and 8 mg/kg possibly due to vasodilatation and/or secretion of the drug into milk. The number of living pups born to rats exposed pre- and postnatally was reduced at 60 mg/kg/day. Based on the results of the pre- and postnatal study, the developmental NOAEL is less than 1 mg/kg/day. Based on plasma exposures in the rat developmental toxicity study, 1mg/kg/day in the pregnant rat is estimated to produce total AUC values for unbound vardenafil and its major metabolite comparable to the human AUC at the MRHD of 20 mg. There are no adequate and well-controlled trials of vardenafil in pregnant women.
Geriatric Use
Elderly males age 65 years and older have higher vardenafil plasma concentrations than younger males (18 - 45 years), mean Cmax and AUC were 34% and 52% higher, respectively (see CLINICAL PHARMACOLOGY, Pharmacokinetics in Special Populations, and DOSAGE AND ADMINISTRATION). Phase 3 clinical trials included more than 834 elderly patients, and no differences in safety or effectiveness of LEVITRA 5, 10, or 20 mg were noted when these elderly patients were compared to younger patients. However, due to increased vardenafil concentrations in the elderly, a starting dose of 5 mg LEVITRA should be considered in patients ≥ 65 years in age.
ADVERSE REACTIONS
LEVITRA was administered to over 4430 men (mean age 56, range 18-89 years; 81% White, 6% Black, 2% Asian, 2% Hispanic and 9% Other) during controlled and uncontrolled clinical trials worldwide. Over 2200 patients were treated for 6 months or longer, and 880 patients were treated for at least 1 year.
In placebo-controlled clinical trials, the discontinuation rate due to adverse events was 3.4% for LEVITRA compared to 1.1% for placebo.
When LEVITRA was taken as recommended in placebo-controlled clinical trials, the following adverse events were reported (see Table 2).
Table 5: Adverse Events Reported By ≥ 2% of Patients Treated with LEVITRA and More Frequent on Drug than Placebo in Fixed and Flexible Dose Randomized, Controlled Trials of 5 mg, 10 mg, or 20 mg Vardenafil
| Adverse Event | Percentage of Patients Reporting Event | |
| Placebo N = 1199 | LEVITRA N = 2203 | |
| Headache | 4% | 15% |
| Flushing | 1% | 11% |
| Rhinitis | 3% | 9% |
| Dyspepsia | 1% | 4% |
| Accidental Injury* | 2% | 3% |
| Sinusitis | 1% | 3% |
| Flu Syndrome | 2% | 3% |
| Dizziness | 1% | 2% |
| Increased Creatine Kinase | 1% | 2% |
| Nausea | 1% | 2% |
| * All the events listed in the above table were deemed to be adverse drug reactions with the exception of accidental injury. | ||
| g Flexible dose studies started all patients at LEVITRA 10 mg and allowed decrease in dose to 5 mg or increase in dose to 20 mg based on side effects and efficacy. | ||
Back pain was reported in 2.0% of patients treated with LEVITRA and 1.7% of patients on placebo.
Placebo-controlled trials suggested a dose effect in the incidence of some adverse events (headache, flushing, dyspepsia, nausea, rhinitis) over the 5 mg, 10 mg, and 20 mg doses of LEVITRA. The following section identifies additional, less frequent events (<2%) reported during the clinical development of LEVITRA. Excluded from this list are those events that are infrequent and minor, those events that may be commonly observed in the absence of drug therapy, and those events that are not reasonably associated with the drug.
Body as a whole: anaphylactic reaction (including laryngeal edema), asthenia, face edema, pain
BODY AS A WHOLE: anaphylactic reaction (including laryngeal edema), asthenia, face edema, pain AUDITORY: tinnitus CARDIOVASCULAR: angina pectoris, chest pain, hypertension, hypotension, myocardial ischemia, myocardial infarction, palpitation, postural hypotension, syncope, tachycardia DIGESTIVE: abdominal pain, abnormal liver function tests, diarrhea, dry mouth, dysphagia, esophagitis, gastritis, gastroesophageal reflux, GGTP increased, vomiting MUSCULOSKELETAL: arthralgia, back pain, myalgia, neck pain NERVOUS: hypertonia, hypesthesia, insomnia, paresthesia, somnolence, vertigo RESPIRATORY: dyspnea, epistaxis, pharyngitis SKIN AND APPENDAGES: photosensitivity reaction, pruritus, rash, sweating OPHTHALMOLOGIC: abnormal vision, blurred vision, chromatopsia, changes in color vision, conjunctivitis (increased redness of the eye), dim vision, eye pain, glaucoma, photophobia, watery eyes UROGENITAL: abnormal ejaculation, priapism (including prolonged or painful erections)
POST-MARKETING EXPERIENCE
Ophthalmologic
Non-arteritic anterior ischemic optic neuropathy (NAION), a cause of decreased vision including permanent loss of vision, has been reported rarely post-marketing in temporal association with the use of phosphodiesterase type 5 (PDE5) inhibitors, including LEVITRA. Most, but not all, of these patients had underlying anatomic or vascular risk factors for development of NAION, including but not necessarily limited to: low cup to disc ratio ("crowded disc"), age over 50, diabetes, hypertension, coronary artery disease, hyperlipidemia and smoking. It is not possible to determine whether these events are related directly to the use of PDE5 inhibitors, to the patient's underlying vascular risk factors or anatomical defects, to a combination of these factors, or to other factors (see PRECAUTIONS/Information for Patients).
Visual disturbances including vision loss (temporary or permanent), such as visual field defect, retinal vein occlusion, and reduced visual acuity, have also been reported rarely in post-marketing experience. It is not possible to determine whether these events are related directly to the use of LEVITRA.
OVERDOSAGE
The maximum dose of LEVITRA for which human data are available is a single 120 mg dose administered to eight healthy male volunteers. The majority of these subjects experienced reversible back pain/myalgia and/or "abnormal vision".
In cases of overdose, standard supportive measures should be taken as required. Renal dialysis is not expected to accelerate clearance because vardenafil is highly bound to plasma proteins and is not significantly eliminated in the urine.
DOSAGE AND ADMINISTRATION
For most patients, the recommended starting dose of LEVITRA is 10 mg, taken orally approximately 60 minutes before sexual activity. The dose may be increased to a maximum recommended dose of 20 mg or decreased to 5 mg based on efficacy and side effects. The maximum recommended dosing frequency is once per day. LEVITRA can be taken with or without food. Sexual stimulation is required for response to treatment.
Geriatrics: A starting dose of 5 mg LEVITRA should be considered in patients ≥ 65 years of age (See CLINICAL PHARMACOLOGY, Pharmacokinetics in Special Populations and PRECAUTIONS).
Hepatic Impairment: For patients with mild hepatic impairment (Child- Pugh A), no dose adjustment of LEVITRA is required. Vardenafil clearance is reduced in patients with moderate hepatic impairment (Child-Pugh B), and a starting dose of 5 mg LEVITRA is recommended. The maximum dose in patients with moderate hepatic impairment should not exceed 10 mg. LEVITRA has not been evaluated in patients with severe hepatic impairment (Child-Pugh C) (see CLINICAL PHARMACOLOGY, Metabolism and Excretion, WARNINGS and PRECAUTIONS).
Renal Impairment: For patients with mild (CLcr = 50-80 ml/min), moderate (CLcr = 30-50 ml/min), or severe (CLcr <30 ml/min) renal impairment, no dose adjustment is required. LEVITRA has not been evaluated in patients on renal dialysis (see CLINICAL PHARMACOLOGY, Metabolism and Excretion and PRECAUTIONS).
Concomitant Medications: The dosage of LEVITRA may require adjustment in patients receiving certain CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, ritonavir, indinavir, and erythromycin) (see WARNINGS, PRECAUTIONS, Drug Interactions). For ritonavir, a single dose of 2.5 mg LEVITRA should not be exceeded in a 72-hour period. For indinavir, ketoconazole 400 mg daily, and itraconazole 400 mg daily, a single dose of 2.5 mg LEVITRA should not be exceeded in a 24-hour period. For ketoconazole 200 mg daily, itraconazole 200 mg daily, and erythromycin, a single dose of 5 mg LEVITRA should not be exceeded in a 24-hour period. For alpha-blockers, caution is advised when PDE5 inhibitors, including LEVITRA, are used concomitantly with alpha-blockers because of the potential for an additive effect on blood pressure. In some patients, concomitant use of these two drug classes can lower blood pressure significantly (see PRECAUTIONS, Alpha-blockers and Drug Interactions) leading to symptomatic hypotension (e.g., fainting). Concomitant treatment should be initiated only if the patient is stable on his alpha blocker therapy. In those patients who are stable on alpha-blocker therapy, LEVITRA should be initiated at a dose of 5 mg (2.5 mg when used concomitantly with certain CYP3A4 inhibitors - see Drug Interactions).
HOW SUPPLIED
LEVITRA (vardenafil HCl) is formulated as orange, film-coated round tablets with debossed "BAYER" cross on one side and "2.5", "5", "10", and "20" on the other side equivalent to 2.5 mg, 5 mg, 10 mg, and 20 mg of vardenafil, respectively.
| Package | Strength | NDC Code |
| Bottles of 30 | 2.5 mg | 0026-8710-69 |
| 5 mg | 0026-8720-69 | |
| 10 mg | 0026-8730-69 | |
| 20 mg | 0026-8740-69 |
Recommended Storage: Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP controlled room temperature].
Bayer Pharmaceuticals Corporation 400 Morgan Lane West Haven, CT 06516 Made in Germany

LEVITRA is a registered trademark of Bayer Aktiengesellschaft and is used under license by GlaxoSmithKline and Schering Corporation.
Continue to
APA Reference
Staff, H.
(2008, December 28). Levitra Full Prescription Information, HealthyPlace. Retrieved
on 2026, April 5 from https://www.healthyplace.com/sex/treatment/levitra-full-prescription-information